How to Evaluate Manufacturer-Provided Extractable Information
Home » How to Evaluate Manufacturer-Provided Extractable Information
Extractables and Leachables (E&L) is a challenging topic to address during product development. A successful E&L program depends on whether the program design is based on the correct information on the container/closure system. The first critical step is the initial material characterization (determination of extractables) of the container/closure system to understand what can possibly migrate into the final drug product as a leachable. If the data from these studies is not applicable or insufficient for a sponsor’s product, it can lead to regulatory delays or recalls. Most of the container/closure systems used today are not proprietary but are purchased from component manufacturers.

As part of a service to their clients, many of these providers now offer extractable information on their products. However, the usefulness of this information can vary widely from manufacturer to manufacturer. The challenge for sponsors is to understand what information they need, what questions to ask their vendors, and how to evaluate the information for their specific application. This presentation will walk through a process for ensuring the right questions are asked and how that information should be evaluated.
Methods: A stepwise process will be presented that will enable companies to ensure they are asking the correct questions when interacting with their container/closure vendors. An example risk assessment and gap analysis process will be presented that covers how the data can be evaluated against a sponsor’s specific drug product.
Results: Representative case studies will be presented in applying the process to data provided by the component manufacturer.
Conclusions: By starting the extractables and leachables evaluation early, sponsors can avoid potential delays in their development time line, avoid recalls and, most importantly, avoid jeopardizing patient safety.
Definitions
Extractable
Compounds that can, under aggressive laboratory conditions, migrate out of materials
Leachable
– Normally a subset of extractables
– Compounds that migrate into the drug product
Extractable ≠ Leachable
– Extractables don’t always leach
– Leachables don’t always extract
Sources of Extractables/Leachables
- Primary packaging components
- Secondary packaging components
- Associated/dosing components
- Processing components
- Shipping materials
Regulatory Basis for Evaluation of Extractables/Leachables
The regulatory requirements for the evaluation of extractables and leachables are found within the Code of Federal Regulations (CFRs), as shown in the tables below. The regulations indicate that the requirements for all contact materials (i.e. final packaging system and manufacturing equipment) are the same.

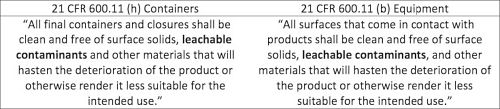
Regulatory Guidance:
Several guidances are available from the FDA which addresses extractables and leachables:
- Guidance for Container Closure Systems for Packaging Human Drugs and Biologics (1999)
- Reviewers Guidance for Nebulizers, Metered Dose Inhalers, Spacers and Actuators (1993)
- Metered Dose Inhalers (MDI) and Dry Powder Inhalers (DPI) (1998)
- Nasal Spray and Inhalers Solution, Suspension, and Spray Drug Products (2002)
- Inhalation Drug Products Packaged in Semipermeable Container Closure Systems (2002)
While the above information from the FDA addresses E&L studies, it doesn’t go into specifics in how the studies need to be performed. For guidance on performing E&L studies industry best practices documents can be used for designing studies. Some of the groups that have guidances available are:
- Product Quality Research Initiative (PQRI)
- Bio-Process Systems Alliance (BPSA)
- Biophorum Operations Group (BPOG)
Additionally, the USP has drafted new chapters, which are currently under review, for guidance on E&L studies:
- <1163> and <1164>
Background
Given the rise in E&L expectations, many component manufacturers have started providing extractable data on their materials. These data packages vary in the level of detail and in how the information was collected. This is not unexpected given the following:
- Lack of regulatory guidance of requirements
- No regulatory requirement for the component manufacturer to perform the studies
- Mainly provided as a sales aid
- Manufacturer does not know all of the possible products and dosing regimens which a company might use their product with
As a result, the usefulness of these packages can vary significantly. It is important for a company to have a process in place to evaluate the information provided by the manufacturer (if any), and to determine whether the information is applicable to their product or whether additional studies will need to be performed. The following is a summary of a detailed process which can be used in walking through some of the main questions to be asked and in how to evaluate the information collected.
Evaluation Process
Step 1: Questions to ask the Vendor
The key to starting the evaluation is to ensure you have asked the correct information and have it available. This requires going to the manufacturer and obtaining as much information as possible on the contact materials, their materials of construction, etc. Below are some example questions which should be posed.
Is there a DMF?
- Drug Master Files (DMFs) can help some level of confidence in the vendors materials. However, it needs to be understood that the FDA does not approve DMF’s. The review them with each new drug which references them. Just because the FDA found the DMF suitable for one use does not necessarily mean it will be found appropriate for another compounds.
- One of the common deficiencies observed in relation to DMF’s is that they are not applicable to the Drug/usage in question.
Is there an extractables data package?
- Some manufacturers have started an extractables program on their own materials and have made these packages available to the client. Some provide a limited package free of charge, while other companies have a package that can be purchased.
- The level of detail and usefulness of this package will vary from vendor to vendor.
Has the component been used in a successful filing?
- This allows for some comfort that the material is being used commercially.
- As with the DMF caveat above, the FDA will review the component in regards to your specific drug product (DP) and application. Just because it was found to be acceptable for another product does not guarantee that the component will be found appropriate for a different product.
Are multiple resin sources available for polymeric components?
- Some manufacturers allow for the use of polymeric resins from multiple sources. This can complicate the extractables profile as each source is unique. Extractables data on each resin will be needed.
Different grades of resins?
- Grades of polymeric resins can have a direct impact on extractables profiles. When choosing a polymeric component, ensure that the decision is based on quality, not just pricing.
Are all components made in the same facility/line?
- If components are made in multiple lines or multiple facilities, then an extractable package should be performed on each material.
What testing is performed on the components for release? i.e. How is variation controlled?
- Does the vendor test the components for extractables as part of a release test.
- These can be important, especially if your product has a high risk for potential leachables. It can help minimize variation in observed peaks.
Will they implement a supply agreement?
- This is critical so that the customer is notified of any changes in the process. Polymeric components are not manufactured in accordance in cGMPs in many cases, as a result more variation can be tolerated.
Step 2: Perform Risk Assessment and Gap Analysis on the information provided.
Once the information from the above questions is received it is important to then evaluate the data in terms of the specific drug product and the specific application. Without a good evaluation process it can leave the company open to higher risk that there could be delays or surprises in the development.
Were chemical extractions performed or list of “possible” extractables given based on manufacturing process?
- Some vendors don’t perform testing on their components, but rather provide information based on the formulation of the component.
- Information based solely on formulation is of limited use, as the specific extractables are often not easily predicted and impurities in the formulation products are not well characterized even though than can often be a source of leachables.
How were the chemical extractions performed?
- A typical extraction profile is as follows:
- Multiple Solvents: Must cover a broad range of solvent polarities, including ones representative of the drug formulation.
- Multiple Extraction Techniques: Should consider the proper extraction techniques to use, such as reflux, microwave, soxhlet., as well as various sample preparation approaches (whole, cut,ground).
- Asymptotic Extractions: Samples are taken from the extractions over time and analyzed to ensure that the maximum level of extractables are being removed.
- Care should be taken to avoid being too aggressive on the extractions, to avoid physically or chemically altering the product being extracted.
- Were the analytical methods used in the analysis appropriate/validated?
- The potential extractables in polymeric material can have a very wide range of chemical properties. It is critical that the analytical methods used are able to cover the full range of potential compounds.
- HPLC – Semi-Volatiles and Non-Volatiles
- GC – Volatiles
- ICP – For metals
- Special Case Extractables
- Poly-aromatic Hydrocarbons (PAH’s)
- Nitrosamines
- 2-Mercaptobenzothiazole
- The potential extractables in polymeric material can have a very wide range of chemical properties. It is critical that the analytical methods used are able to cover the full range of potential compounds.
Did the analytical methods go low enough?
- This is the most common shortcoming of data packages provided by manufacturers. See the following section for calculation of the Analytical Evaluation Threshold (AET) and the Case Study examples
Evaluation of Supply/Shipping chain
- While very rare, shipping materials have been known to be the source of leachables. Additionally, how the materials are packaged and shipped from the manufacturer are important to understand.
Commonly Observed Gaps/Deficiencies
- Solvent(s) used in extraction studies not representative of the Drug Product formulation
- Asymptotic extractions were not performed
- Reporting level of the data is higher than the determined AET.
- Insufficient number of analytical methods used in analysis of extracts.
- Inadequate detail in reporting extractables
- Compounds not properly identified, e.g. “phosphite based anti-oxidant” or “proprietary curing agent”
- Compounds named based upon formulation rather than actual identity, e.g. silicone oil
How Low To Go
One of the most challenging and common deficiencies is that the analytical methods didn’t go low enough for a specific drug products formulation/dosing regimen.
No direct regulatory guidance is available
- ICH guidelines for general impurities do not apply
- Genotoxic Impurities Guidance – 1.5 µg/day
- Ophthalmics1 – 1 ppm reporting, 10 ppm Identification, 20 ppm Qualification
Safety Concern Threshold (SCT)
- Level below which there is negligible risk associated with the toxicity of the extractable/leachable compound, based upon dosing
- Only applies to unknowns
- Presented as a Total Daily Intake (TDI): usually in µg/day
- Product Quality Research Institute (PQRI) recommends 0.15 µg/day for inhalation products
- Current thinking of PQRI Parenteral and Ophthalmic Drug Product (PODP) working group is 1.5 µg/day for parenteral products
Using the PQRI’s idea of the Safety Concern Threshold we can convert that to an equivalent analytical level for a specific product. The conversion takes into account the drug’s specific dosing regimen and the number of doses which are in each container/closure system. Example equations for converting the AET into a number of different units, depending on the usage, are shown below.

1Ng, Linda (CDER/FDA), “Current Regulatory Recommendations for Leachables in Ophthalmic Drug Products,” Thresholds and Best Practices for Parenteral and Ophthalmic Drug Products, 22-23 February 2011, Bethesda, MD.
Case Study #1
One of the most critical issues in extractable evaluations is ensuring that the data you are making the decision meets the expectations based on the best practices recommendations. This requires evaluating the information for the specific drug product formulation and worst case scenario dosing regimen for a product. The following case study demonstrates that information provided by a manufacturer may be adequate for some cases but not acceptable for others.
The product is a parenteral with an aqueous formulation (same formulation for both scenarios) in a 20 mm glass vial with a 3.0 gram rubber stopper. The stopper manufacturer has provided an extractables package that used analytical methods with a quantitation limit (QL) of 5ppm. Asymptotic extractions were performed using water and IPA.
We will review the package against two different container closure scenarios:
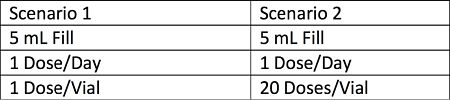
For the evaluation of the data we are going to examine two specific items:
1. Extraction Solvent(s) vs. DP formulation
– The extraction solvents used (water and IPA) cover the polarity range of the DP formulation (aqueous) used above. In terms of extraction solvents used, the data provided by the manufacturer are applicable to the product.
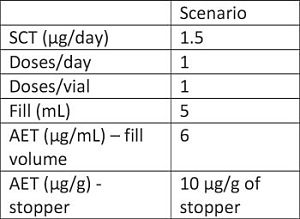
2. QL of the extraction methods vs. AET needed
– Applying a 1.5 µg/day SCT, based on the PQRI current thinking for parenterals, we are able to compare the expected reporting level against the data provided by the manufacturer.
As can be seen in the table, for scenario 2, we are able to use the data provided by the manufacturer for evaluating the extractables profiles, however in the case of scenario 1, the analytical methods used didn’t go low enough.
Case Study #1 summary: The data provided by the manufacturer can be used for the evaluation in scenario 2. However, the data are not adequate for evaluating against the dosing regimen in scenario 1. As a result, it is likely that additional studies would be needed to achieve the lower QL required by the AET.
Case Study #2
For this example we will evaluate another parenteral product with an oil based formulation (cottonseed oil) in the same container/closure system as in Case Study #1. The stopper manufacturer has provided an extractables package that used analytical methods with a quantitation limit of 5ppm. Asymptotic extractions were performed using water and IPA.
We will review the package against the following configuration:

For the evaluation of the data we are going to evaluate the same two items as we did in case study #1:
1. Extraction Solvent(s) vs. DP formulation
– The extraction solvents (water and IPA) are not representative, nor do they cover the polarity range of the formulation(cottonseed oil). As a result they would not be predictive of leachables in the product.
2. QL of the extraction methods vs. AET needed
– Applying a 1.5 ug/day SCT, based on the PQRI current thinking for parenterals, we are able to compare the expected reporting level against the data provided by the manufacturer.
As can be seen in the table, the QL (5ppm) and extractables data provided by the manufacturer does go low enough compared to the AET required for the drug product.
Case Study #2 summary: Based on the extraction solvent mis-match, the data provided by the manufacturer cannot be used in evaluation for the drug product. New studies would be required.
Summary
It is important to use the correct information and process to ensure that the development process is not delayed as a result of an E&L issue. By having the right information available, it is possible to avoid some of the most common pitfalls that can result in regulatory delays for products by having a well-defined process in place when choosing the container/closure system.
Would you like to learn more about evaluating Manufacturer Extractable Information?
Contact us today to evaluate manufacturer extractable information. Please complete the form below to have an EAG expert contact you.